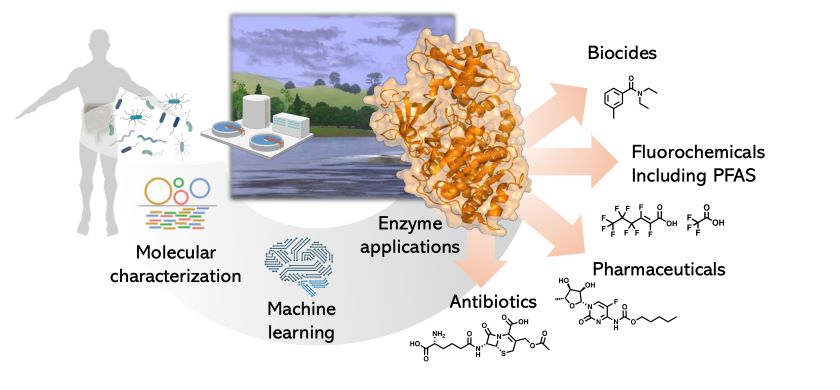
Unsere Forschungsgruppe untersucht, wie natürlich vorkommende Mikroben, einschliesslich Bakterien und Pilzen, chemische Schadstoffe abbauen können. Wir nutzen eine Kombination aus Experimenten und computergestützter Modellierung, um vorherzusagen, wie unterschiedliche mikrobielle Enzyme Industriechemikalien, Pharmazeutika und Agrochemikalien umwandeln.
Ein zentrales Thema ist der Fokus auf fluorierte Verbindungen, insbesondere PFAS (Per- und Polyfluoralkylsubstanzen), die aufgrund ihrer Persistenz in der Umwelt als "ewige Chemikalien" bezeichnet werden. Ein Grund für ihre Abbauresistenz ist die starke chemische Kohlenstoff-Fluor-Bindung. Wir konnten zeigen, dass einige Mikroben, darunter auch Bakterien aus dem menschlichen Darm, Enzyme besitzen, die C-F-Bindungen spalten können. Allerdings sind diese Enzyme nur bei einfachen fluorierten Verbindungen mit nur einem oder zwei Fluoratomen wirksam, nicht jedoch bei langkettigen, perfluorierten PFAS, die oft fünfzehn oder mehr Fluoratome enthalten. Wir arbeiten daran, diese Aktivität zu verbessern und herauszufinden, wie die enzymatische Vielfalt von Mikroorganismen die große chemische Diversität fluorierter Verbindungen bewältigen kann. Unser langfristiges Ziel ist es, unsere Fähigkeit zu verbessern, mikrobielle Enzyme zur Erkennung und Entfernung von Schadstoffen zu verstehen, vorherzusagen und zu entwickeln.
Ein weiteres wichtiges Forschungsfeld in unserer Gruppe ist die mikrobielle Biosynthese. Hier interessieren uns mikrobielle Bioprodukte wie Sekundärmetaboliten und extrazelluläre polymere Substanzen sowie deren ökologische Funktionen.
Für weitere Informationen können Sie sich gerne direkt an uns wenden!
Für eine vollständige Publikationsliste siehe: Google Scholar
array(4 items)0 => Snowflake\Publications\Domain\Model\Publicationprototypepersistent entity (uid=33689, pid=124)originalId => protected33689 (integer)
authors => protected'Wackett, L. P.; Robinson, S. L.' (51 chars)
title => protected'A prescription for engineering PFAS biodegradation' (50 chars)
journal => protected'Biochemical Journal' (19 chars)
year => protected2024 (integer)
volume => protected481 (integer)
issue => protected'23' (2 chars)
startpage => protected'1757' (4 chars)
otherpage => protected'1770' (4 chars)
categories => protected'' (0 chars)
description => protected'Per- and polyfluorinated chemicals (PFAS) are of rising concern due to envir onmental persistence and emerging evidence of health risks to humans. Enviro nmental persistence is largely attributed to a failure of microbes to degrad e PFAS. PFAS recalcitrance has been proposed to result from chemistry, speci fically C-F bond strength, or biology, largely negative selection from fluor ide toxicity. Given natural evolution has many hurdles, this review advocate s for a strategy of laboratory engineering and evolution. Enzymes identified to participate in defluorination reactions have been discovered in all Enzy me Commission classes, providing a palette for metabolic engineering. In viv o PFAS biodegradation will require multiple types of reactions and powerful fluoride mitigation mechanisms to act in concert. The necessary steps are to : (1) engineer bacteria that survive very high, unnatural levels of fluoride , (2) design, evolve, and screen for enzymes that cleave C-F bonds in a broa der array of substrates, and (3) create overall physiological conditions tha t make for positive selective pressure with PFAS substrates.' (1124 chars)
serialnumber => protected'0264-6021' (9 chars)
doi => protected'10.1042/BCJ20240283' (19 chars)
uid => protected33689 (integer)
_localizedUid => protected33689 (integer)modified_languageUid => protectedNULL
_versionedUid => protected33689 (integer)modifiedpid => protected124 (integer)1 => Snowflake\Publications\Domain\Model\Publicationprototypepersistent entity (uid=34896, pid=124)originalId => protected34896 (integer)
authors => protected'Probst, S. I.; Felder, F. D.; Poltorak, V.; Mewalal , R.; Blaby, I. K.; Robinson, S. L.' (136 chars)
title => protected'Enzymatic carbon–fluorine bond cleavage by human gut microbes' (63 chars)
journal => protected'Proceedings of the National Academy of Sciences of the United States of Amer ica PNAS' (84 chars)
year => protected2025 (integer)
volume => protected122 (integer)
issue => protected'24' (2 chars)
startpage => protected'e2504122122 (12 pp.)' (20 chars)
otherpage => protected'' (0 chars)
categories => protected'human gut microbiome; defluorination; haloacid dehalogenases; molecular dyna mics; protein engineering' (101 chars)
description => protected'Fluorinated compounds are used for agrochemical, pharmaceutical, and numerou s industrial applications, resulting in global contamination. In many molecu les, fluorine is incorporated to enhance the half-life and improve bioavaila bility. Fluorinated compounds enter the human body through food, water, and xenobiotics including pharmaceuticals, exposing gut microbes to these substa nces. The human gut microbiota is known for its xenobiotic biotransformation capabilities, but it was not previously known whether gut microbial enzymes could break carbon–fluorine bonds, potentially altering the toxicity of t hese compounds. Here, through the development of a rapid, miniaturized fluor ide detection assay for whole-cell screening, we identified active gut micro bial defluorinases. We biochemically characterized enzymes from diverse huma n gut microbial classes including Clostridia, Bacilli, and Coriobacteriia, w ith the capacity to hydrolyze (di)fluorinated organic acids and a fluorinate d amino acid. Whole-protein alanine scanning, molecular dynamics simulations , and chimeric protein design enabled the identification of a disordered C-t erminal protein segment involved in defluorination activity. Domain swapping exclusively of the C-terminus conferred defluorination activity to a nondef luorinating dehalogenase. To advance our understanding of the structural and sequence differences between defluorinating and nondefluorinating dehalogen ases, we trained machine learning models which identified protein termini as important features. Models trained on 41-amino acid segments from protein C termini alone predicted defluorination activity with 83% accuracy (compared to 95% accuracy based on full-length protein features). This work is releva nt for therapeutic interventions and environmental and human health by uncov ering specificity-determining signatures of fluorine biochemistry from the g ut microbiome.' (1914 chars)
serialnumber => protected'0027-8424' (9 chars)
doi => protected'10.1073/pnas.2504122122' (23 chars)
uid => protected34896 (integer)
_localizedUid => protected34896 (integer)modified_languageUid => protectedNULL
_versionedUid => protected34896 (integer)modifiedpid => protected124 (integer)2 => Snowflake\Publications\Domain\Model\Publicationprototypepersistent entity (uid=33317, pid=124)originalId => protected33317 (integer)
authors => protected'Attrah, M.; Schärer, M. R.; Esposito, M.; Gionchetta,&n bsp;G.; Bürgmann, H.; Lens, P. N. L.; Fenner, K.; van de Vossenberg, J.; Robinson, S. L.' (205 chars)
title => protected'Disentangling abiotic and biotic effects of treated wastewater on stream bio film resistomes enables the discovery of a new planctomycete beta-lactamase' (151 chars)
journal => protected'Microbiome' (10 chars)
year => protected2024 (integer)
volume => protected12 (integer)
issue => protected'' (0 chars)
startpage => protected'164 (15 pp.)' (12 chars)
otherpage => protected'' (0 chars)
categories => protected'antibiotic resistance genes; stream biofilms; metagenomics; wastewater efflu ent; sulfonamides; planctomycetota; beta-lactamases' (127 chars)
description => protected'<em>Background</em> Environmental reservoirs of antibiotic resistance pose a threat to human and animal health. Aquatic biofilms impacted by wastewater effluent (WW) are known environmental reservoirs for antibiotic resistance; however, the relative importance of biotic factors and abiotic factors from WW on the abundance of antibiotic resistance genes (ARGs) within aquatic bi ofilms remains unclear. Additionally, experimental evidence is limited withi n complex aquatic microbial communities as to whether genes bearing low sequ ence similarity to validated reference ARGs are functional as ARGs.<br /><br /><em>Results</em> To disentangle the effects of abiotic and biotic factors on ARG abundances, natural biofilms were previously grown in flume systems with different proportions of stream water and either ultrafiltered or non-u ltrafiltered WW. In this study, we conducted deep shotgun metagenomic sequen cing of 75 biofilm, stream, and WW samples from these flume systems and comp ared the taxonomic and functional microbiome and resistome composition. Stat istical analysis revealed an alignment of the resistome and microbiome compo sition and a significant association with experimental treatment. Several AR G classes exhibited an increase in normalized metagenomic abundances in biof ilms grown with increasing percentages of non-ultrafiltered WW. In contrast, sulfonamide and extended-spectrum beta-lactamase ARGs showed greater abunda nces in biofilms grown in ultrafiltered WW compared to non-ultrafiltered WW. Overall, our results pointed toward the dominance of biotic factors over ab iotic factors in determining ARG abundances in WW-impacted stream biofilms a nd suggested gene family-specific mechanisms for ARGs that exhibited diverge nt abundance patterns. To investigate one of these specific ARG families exp erimentally, we biochemically characterized a new beta-lactamase from the <e m>Planctomycetota</em> (<em>Phycisphaeraceae</em>). This beta-lactamase disp layed activity in the cl...' (2583 chars)
serialnumber => protected'2049-2618' (9 chars)
doi => protected'10.1186/s40168-024-01879-w' (26 chars)
uid => protected33317 (integer)
_localizedUid => protected33317 (integer)modified_languageUid => protectedNULL
_versionedUid => protected33317 (integer)modifiedpid => protected124 (integer)3 => Snowflake\Publications\Domain\Model\Publicationprototypepersistent entity (uid=33730, pid=124)originalId => protected33730 (integer)
authors => protected'Marti, T. D.; Schweizer, D.; Yu, Y.; Schärer, M.&n bsp;R.; Probst, S. I.; Robinson, S. L.' (134 chars)
title => protected'Machine learning reveals signatures of promiscuous microbial amidases for mi cropollutant biotransformations' (107 chars)
journal => protected'ACS Environmental Au' (20 chars)
year => protected2025 (integer)
volume => protected5 (integer)
issue => protected'1' (1 chars)
startpage => protected'114' (3 chars)
otherpage => protected'127' (3 chars)
categories => protected'micropollutant biotransformations; amidase signature enzymes; urinary microb iota; paracetamol; acetylsulfamethoxazole; capecitabine; machine learning' (149 chars)
description => protected'Organic micropollutants, including pharmaceuticals, personal care products, pesticides, and food additives, are widespread in the environment, causing p otentially toxic effects. Human waste is a direct source of micropollutants, with the majority of pharmaceuticals being excreted through urine. Urine co ntains its own microbiota with the potential to catalyze micropollutant biot ransformations. Amidase signature (AS) enzymes are known for their promiscuo us activity in micropollutant biotransformations, but the potential for AS e nzymes from the urinary microbiota to transform micropollutants is not known . Moreover, the characterization of AS enzymes to identify key chemical and enzymatic features associated with biotransformation profiles is critical fo r developing benign-by-design chemicals and micropollutant removal strategie s. Here, to uncover the signatures of AS enzyme-substrate specificity, we te sted 17 structurally diverse compounds against a targeted enzyme library con sisting of 40 AS enzyme homologues from diverse urine microbial isolates. Th e most promiscuous enzymes were active on nine different substrates, while 1 6 enzymes had activity on at least one substrate and exhibited diverse subst rate specificities. Using an interpretable gradient boosting machine learnin g model, we identified chemical and amino acid features associated with AS e nzyme biotransformations. Key chemical features from our substrates included the molecular weight of the amide carbonyl substituent and the number of fo rmal charges in the molecule. Four of the identified amino acid features wer e located in close proximity to the substrate tunnel entrance. Overall, this work highlights the understudied potential of urine-derived microbial AS en zymes for micropollutant biotransformation and offers insights into substrat e and protein features associated with micropollutant biotransformations for future environmental applications.' (1935 chars)
serialnumber => protected'' (0 chars)
doi => protected'10.1021/acsenvironau.4c00066' (28 chars)
uid => protected33730 (integer)
_localizedUid => protected33730 (integer)modified_languageUid => protectedNULL
_versionedUid => protected33730 (integer)modifiedpid => protected124 (integer)
A prescription for engineering PFAS biodegradation
Per- and polyfluorinated chemicals (PFAS) are of rising concern due to environmental persistence and emerging evidence of health risks to humans. Environmental persistence is largely attributed to a failure of microbes to degrade PFAS. PFAS recalcitrance has been proposed to result from chemistry, specifically C-F bond strength, or biology, largely negative selection from fluoride toxicity. Given natural evolution has many hurdles, this review advocates for a strategy of laboratory engineering and evolution. Enzymes identified to participate in defluorination reactions have been discovered in all Enzyme Commission classes, providing a palette for metabolic engineering. In vivo PFAS biodegradation will require multiple types of reactions and powerful fluoride mitigation mechanisms to act in concert. The necessary steps are to: (1) engineer bacteria that survive very high, unnatural levels of fluoride, (2) design, evolve, and screen for enzymes that cleave C-F bonds in a broader array of substrates, and (3) create overall physiological conditions that make for positive selective pressure with PFAS substrates.
Enzymatic carbon–fluorine bond cleavage by human gut microbes
Fluorinated compounds are used for agrochemical, pharmaceutical, and numerous industrial applications, resulting in global contamination. In many molecules, fluorine is incorporated to enhance the half-life and improve bioavailability. Fluorinated compounds enter the human body through food, water, and xenobiotics including pharmaceuticals, exposing gut microbes to these substances. The human gut microbiota is known for its xenobiotic biotransformation capabilities, but it was not previously known whether gut microbial enzymes could break carbon–fluorine bonds, potentially altering the toxicity of these compounds. Here, through the development of a rapid, miniaturized fluoride detection assay for whole-cell screening, we identified active gut microbial defluorinases. We biochemically characterized enzymes from diverse human gut microbial classes including Clostridia, Bacilli, and Coriobacteriia, with the capacity to hydrolyze (di)fluorinated organic acids and a fluorinated amino acid. Whole-protein alanine scanning, molecular dynamics simulations, and chimeric protein design enabled the identification of a disordered C-terminal protein segment involved in defluorination activity. Domain swapping exclusively of the C-terminus conferred defluorination activity to a nondefluorinating dehalogenase. To advance our understanding of the structural and sequence differences between defluorinating and nondefluorinating dehalogenases, we trained machine learning models which identified protein termini as important features. Models trained on 41-amino acid segments from protein C termini alone predicted defluorination activity with 83% accuracy (compared to 95% accuracy based on full-length protein features). This work is relevant for therapeutic interventions and environmental and human health by uncovering specificity-determining signatures of fluorine biochemistry from the gut microbiome.
Probst, S. I.; Felder, F. D.; Poltorak, V.; Mewalal, R.; Blaby, I. K.; Robinson, S. L. (2025) Enzymatic carbon–fluorine bond cleavage by human gut microbes, Proceedings of the National Academy of Sciences of the United States of America PNAS, 122(24), e2504122122 (12 pp.), doi:10.1073/pnas.2504122122, Institutional Repository
Disentangling abiotic and biotic effects of treated wastewater on stream biofilm resistomes enables the discovery of a new planctomycete beta-lactamase
Background Environmental reservoirs of antibiotic resistance pose a threat to human and animal health. Aquatic biofilms impacted by wastewater effluent (WW) are known environmental reservoirs for antibiotic resistance; however, the relative importance of biotic factors and abiotic factors from WW on the abundance of antibiotic resistance genes (ARGs) within aquatic biofilms remains unclear. Additionally, experimental evidence is limited within complex aquatic microbial communities as to whether genes bearing low sequence similarity to validated reference ARGs are functional as ARGs.
Results To disentangle the effects of abiotic and biotic factors on ARG abundances, natural biofilms were previously grown in flume systems with different proportions of stream water and either ultrafiltered or non-ultrafiltered WW. In this study, we conducted deep shotgun metagenomic sequencing of 75 biofilm, stream, and WW samples from these flume systems and compared the taxonomic and functional microbiome and resistome composition. Statistical analysis revealed an alignment of the resistome and microbiome composition and a significant association with experimental treatment. Several ARG classes exhibited an increase in normalized metagenomic abundances in biofilms grown with increasing percentages of non-ultrafiltered WW. In contrast, sulfonamide and extended-spectrum beta-lactamase ARGs showed greater abundances in biofilms grown in ultrafiltered WW compared to non-ultrafiltered WW. Overall, our results pointed toward the dominance of biotic factors over abiotic factors in determining ARG abundances in WW-impacted stream biofilms and suggested gene family-specific mechanisms for ARGs that exhibited divergent abundance patterns. To investigate one of these specific ARG families experimentally, we biochemically characterized a new beta-lactamase from the Planctomycetota (Phycisphaeraceae). This beta-lactamase displayed activity in the cleavage of cephalosporin analog despite sharing a low sequence identity with known ARGs.
Conclusions This discovery of a functional planctomycete beta-lactamase ARG is noteworthy, not only because it was the first beta-lactamase to be biochemically characterized from this phylum, but also because it was not detected by standard homology-based ARG tools. In summary, this study conducted a metagenomic analysis of the relative importance of biotic and abiotic factors in the context of WW discharge and their impact on both known and new ARGs in aquatic biofilms.
Attrah, M.; Schärer, M. R.; Esposito, M.; Gionchetta, G.; Bürgmann, H.; Lens, P. N. L.; Fenner, K.; van de Vossenberg, J.; Robinson, S. L. (2024) Disentangling abiotic and biotic effects of treated wastewater on stream biofilm resistomes enables the discovery of a new planctomycete beta-lactamase, Microbiome, 12, 164 (15 pp.), doi:10.1186/s40168-024-01879-w, Institutional Repository
Machine learning reveals signatures of promiscuous microbial amidases for micropollutant biotransformations
Organic micropollutants, including pharmaceuticals, personal care products, pesticides, and food additives, are widespread in the environment, causing potentially toxic effects. Human waste is a direct source of micropollutants, with the majority of pharmaceuticals being excreted through urine. Urine contains its own microbiota with the potential to catalyze micropollutant biotransformations. Amidase signature (AS) enzymes are known for their promiscuous activity in micropollutant biotransformations, but the potential for AS enzymes from the urinary microbiota to transform micropollutants is not known. Moreover, the characterization of AS enzymes to identify key chemical and enzymatic features associated with biotransformation profiles is critical for developing benign-by-design chemicals and micropollutant removal strategies. Here, to uncover the signatures of AS enzyme-substrate specificity, we tested 17 structurally diverse compounds against a targeted enzyme library consisting of 40 AS enzyme homologues from diverse urine microbial isolates. The most promiscuous enzymes were active on nine different substrates, while 16 enzymes had activity on at least one substrate and exhibited diverse substrate specificities. Using an interpretable gradient boosting machine learning model, we identified chemical and amino acid features associated with AS enzyme biotransformations. Key chemical features from our substrates included the molecular weight of the amide carbonyl substituent and the number of formal charges in the molecule. Four of the identified amino acid features were located in close proximity to the substrate tunnel entrance. Overall, this work highlights the understudied potential of urine-derived microbial AS enzymes for micropollutant biotransformation and offers insights into substrate and protein features associated with micropollutant biotransformations for future environmental applications.
Marti, T. D.; Schweizer, D.; Yu, Y.; Schärer, M. R.; Probst, S. I.; Robinson, S. L. (2025) Machine learning reveals signatures of promiscuous microbial amidases for micropollutant biotransformations, ACS Environmental Au, 5(1), 114-127, doi:10.1021/acsenvironau.4c00066, Institutional Repository
Projekte
EXPLORA kartiert ökologische Muster extremer aquatischer Vielfalt, um neue Enzyme und Metaboliten zu identifizieren.
Ziel dieses Projekts ist die Charakterisierung, Modellierung und Vorhersage von Enzymfamilien, die die Biotransformation von Schadstoffen in Periphyton vorantreiben.